

Non-Conformances, CAPA, and Change Control: Three Tools Every Quality Team Needs to Understand
What Is a Non-Conformance?
A non-conformance occurs when something does not meet a defined requirement. That requirement might come from a product specification, a standard operating procedure (SOP), or a regulatory expectation such as 21 CFR Part 211.
Common examples include:
- A batch that fails testing (out-of-specification, or OOS, result)
- An SOP that was not followed during manufacturing or testing
- An incorrect label applied to a product
- A temperature excursion during storage or shipping
- A documentation error discovered during review
- An equipment failure that impacts a process
Non-conformances do not exist in isolation. Related event types like deviations, Out-of-Specification (OOS) results, and Out-of-Trend (OOT) results represent distinct signals that the system is not performing as intended, and each carries its own investigation requirements and documentation obligations.
Terminology can vary by industry: pharmaceutical and biotech organizations lean on “deviation” and “OOS,” medical device companies use “nonconformance” per ISO 13485, and food and beverage operations refer to “defects” or “nonconformities” under ISO 9001. Regardless of the label, any of these events has the potential to escalate into a corrective and preventive action (CAPA).
The primary purpose of a non-conformance record is straightforward: document what happened and contain the risk. That might mean placing product on hold, quarantining materials, or halting a process while the issue is evaluated. The non-conformance is the record of the event but it is not the investigation.
What Is a CAPA?
A corrective and preventive action (CAPA) is the structured investigation process used to identify the root cause of a problem, correct it, and prevent it from recurring. CAPAs can be opened in response to a non-conformance (reactive), or they can be initiated without one (proactive).
Reactive CAPA is triggered by:
- A confirmed non-conformance
- Repeated issues or negative trends in quality data
- Audit findings or FDA Form 483 observations
Proactive CAPA is triggered by:
- Trend analysis showing an increase in minor deviations before a major failure occurs
- An internal audit that identifies a system gap
- A pattern emerging in customer complaint data
- A risk assessment that surfaces a process vulnerability
The distinction matters: a CAPA is not just a corrective action, it is a documented investigation with root cause analysis as its foundation. Closing a CAPA without adequately addressing the root cause is one of the most common findings during FDA inspections and third-party audits.
What Is Change Control?
Change control is a structured process for evaluating and implementing planned changes in a controlled, documented manner. It is most commonly used for proactive improvements.
Examples of changes that require a change control:
- Qualifying a new supplier or raw material
- Revising an existing SOP
- Modifying equipment or manufacturing processes
- Implementing process improvements identified through trend analysis
Change control exists to assess risk before a change is made and to ensure that the change does not inadvertently introduce new compliance risk or product quality issues. In many cases, change control is used entirely on its own, there is no non-conformance and no CAPA involved.
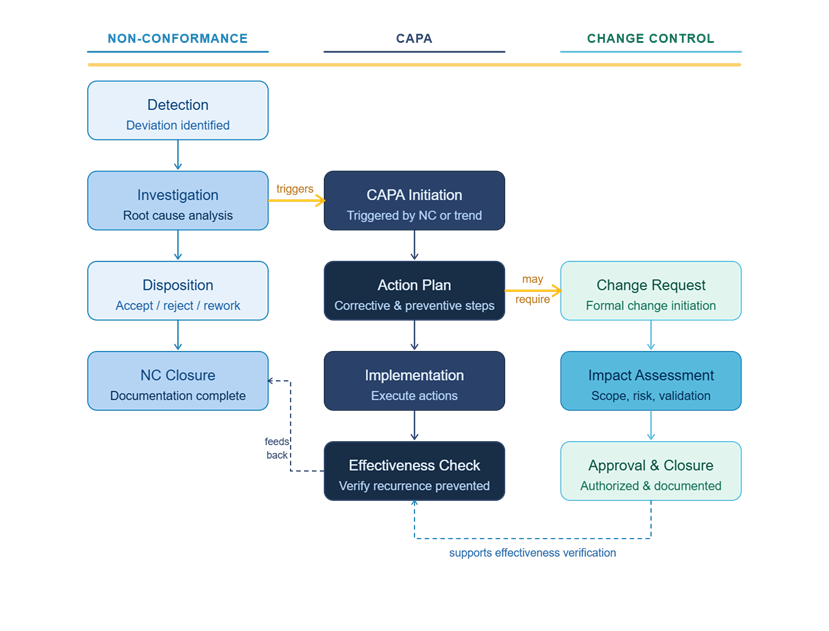
How These Three Tools Work Together
Understanding each tool individually is important, but the real value comes from knowing how they interact in practice.
Reactive flow: A non-conformance occurs. The quality team evaluates severity and determines whether the issue requires a formal investigation. If it does, a CAPA is opened. If the CAPA investigation determines that a process, procedure, or system change is required to prevent recurrence, that change is implemented through change control.

Proactive flow: Trend data or a risk assessment identifies a potential vulnerability before a failure occurs. A CAPA is opened to address the systemic risk. The solution identified through the CAPA is implemented through change control.

In both flows, each tool plays a distinct role. Non-conformance documents the event. CAPA drives the investigation and corrective strategy. Change control manages the implementation of the solution.
Common Mistakes to Avoid
Even experienced quality professionals occasionally conflate these tools. Here are the most common errors:
Using CAPA for routine changes. Not every change requires a CAPA. If you are updating an SOP because a process improved — not because something went wrong — that is a change control, not a CAPA.
Using change control to fix a problem without investigating it first. If a non-conformance occurred and a change is clearly needed, it can be tempting to simply open a change control and move on. However, skipping the CAPA means skipping the root cause investigation and the same problem is likely to recur.
Failing to document non-conformances. Every deviation from a requirement should be documented, even if it appears minor. Undocumented non-conformances create gaps in your quality record and can obscure trends that would otherwise be visible.
Treating CAPA as only reactive. Proactive CAPAs are a sign of a mature quality system. Regulators and auditors look favorably on organizations that identify and address systemic risk before it becomes a failure.
The Bottom Line
Non-conformances, CAPAs, and change controls serve specific functions within a GMP-compliant Quality Management System. Used correctly and in the right sequence, they form a system that not only protects product quality but also supports regulatory compliance and reduces the likelihood of repeat occurrences.
Frequently Asked Questions
The terms are often used interchangeably, but in regulated pharmaceutical and biotech environments, a deviation typically refers to an unplanned departure from an approved procedure or process during manufacturing or testing, while a non-conformance refers more broadly to any failure to meet a defined requirement including product specifications, labeling standards, or regulatory expectations. Medical device companies operating under ISO 13485 tend to use “nonconformance” as the primary term. Regardless of terminology, both require documentation, evaluation of impact, and a determination of whether further investigation is warranted.
No. A non-conformance documents the event and contains the immediate risk. Whether it escalates to a CAPA depends on the severity of the issue, its potential impact on product quality or patient safety, and whether the root cause is known and simple to address. A single, isolated minor deviation with an obvious cause and a straightforward correction may not require a formal CAPA. However, recurring non-conformances, issues with significant quality or regulatory impact, and events that cannot be fully explained should always trigger a CAPA. Your quality procedures should define criteria for CAPA escalation so that the decision is documented and defensible during an FDA inspection.
The most widely used methods in FDA-regulated industries include the 5 Whys, fishbone (Ishikawa) diagrams, fault tree analysis, and failure mode and effects analysis (FMEA). For straightforward issues, the 5 Whys provides a structured path from symptom to systemic cause. Fishbone diagrams are useful when the cause is not immediately apparent and multiple contributing factors need to be evaluated across categories such as people, process, equipment, materials, environment, and measurement. The method you select should be appropriate to the complexity of the issue and should be documented in the CAPA record. Regulators expect to see evidence of genuine investigation not a conclusion written before the analysis was performed.
There is no universal regulatory timeframe, but most quality systems define target closure timelines internally commonly 30 days for minor CAPAs and 60 to 90 days for more complex investigations. What matters to FDA investigators is not just the duration but whether there is documented justification for any extension and evidence that the CAPA is progressing. A large backlog of aged, open CAPAs is consistently cited as a systemic quality failure during inspections. Build your CAPA procedure to require periodic status reviews, owner accountability, and documented rationale whenever a target date is extended.
Any planned change that has the potential to affect product quality, process performance, regulatory status, or validated state should go through change control. This includes revisions to SOPs and batch records, changes to raw material suppliers or specifications, equipment modifications or replacements, facility changes, software updates to systems used in GMP operations, and alterations to validated processes or analytical methods. A well-designed change control procedure includes a risk assessment step to classify the change, identify required pre-implementation activities such as validation or regulatory notification, and define the approval chain before the change is executed.
Effectiveness verification is the step most often skipped or superficially completed, and it is one of the first things FDA investigators look for. Verification should be defined upfront when the CAPA is opened, before corrective actions are implemented, and should specify what evidence will be collected, over what time period, and what threshold constitutes success. For example, if a CAPA was opened because a particular deviation occurred three times in six months, the effectiveness check might monitor for recurrence over the following six months following implementation of corrective actions. The CAPA should not be closed until that evidence has been collected and reviewed, and the conclusion documented.
Yes, and this is actually the most common scenario in a mature quality system. A non-conformance occurs, a CAPA investigation identifies the root cause, and the corrective action requires a change to a procedure, process, or system. That change is then implemented through change control. The CAPA and change control records should be cross-referenced so that the full chain of events, from the original failure through the investigation to the implemented solution, is traceable in a single audit trail. This linkage is important during inspections and provides clear evidence that the organization identified, investigated, resolved, and documented the issue systematically.
Start with your non-conformance system, because it is the foundation for everything else. You cannot run a CAPA program without a reliable mechanism to capture quality events, and you cannot make informed change control decisions without data from your non-conformance history. Write a clear, simple procedure that defines what constitutes a non-conformance in your operation, who is responsible for initiating a record, and what the minimum documentation requirements are. Once that is in place, layer in your CAPA procedure with defined escalation criteria and a straightforward investigation workflow. Change control can follow. The goal at the outset is not a perfect system, it is a working system that captures events, drives investigation, and produces documented evidence of resolution. You can optimize as you mature.
cGMP Consulting provides engineering, regulatory compliance, and quality systems support to pharmaceutical, biotechnology, and other FDA-regulated manufacturers. Our consultants bring direct experience with QMS implementation, FDA inspection readiness, and CAPA program development across a wide range of regulated environments.
Interested in learning more about quality systems? Visit us at cGMPConsulting.com.
Author:
Tamara Neumann is a Project Manager at cGMP Consulting with strong expertise in regulatory compliance, particularly for the dietary supplement and cosmetics industries. She has a proven track record of guiding organizations through the design and implementation of robust Quality Management Systems, enabling clients to achieve and sustain GMP compliance with FDA regulations and applicable industry standards. Her hands-on approach and familiarity with regulatory requirements have made her a trusted partner for companies looking to strengthen their compliance programs.




{kind=link}
{kind=link}